Executing the Program¶
The main workflow is executed through pyEpiAneufinder.epiAneufinder().
When using a fragment file as input, the file should be ordered first by cell
barcode and then by genomic position. pyEpiAneufinder can do this sorting
internally when sort_fragment=True.
Alternatively, the fragment_file argument can point to a Cell Ranger-style
matrix directory containing the peaks, barcodes, and matrix files. In that
case, set cellRangerInput=True. Fragment-file input is still the preferred
path when available.
By default, the workflow performs GC correction. The correction can be disabled
with GC=False, but the default is recommended.
The workflow also needs:
a reference genome FASTA file
a blacklist BED file
a window size
an output directory
optional chromosomes to exclude
a minimum fragment cutoff for filtering cells
For example:
import pyEpiAneufinder as pea
pea.epiAneufinder(
fragment_file="sample_data/sample.tsv.gz",
outdir="results_sample_data",
genome_file="hg38.fa.gz",
blacklist="sample_data/hg38-blacklist.v2.bed",
windowSize=100000,
ncores=1,
exclude=["chrX", "chrY"],
minFrags=20000,
resume=False,
cellRangerInput=False,
GC=True,
sort_fragment=True,
remove_barcodes=None,
)
The test dataset used in the examples is available in the repository under
sample_data/.
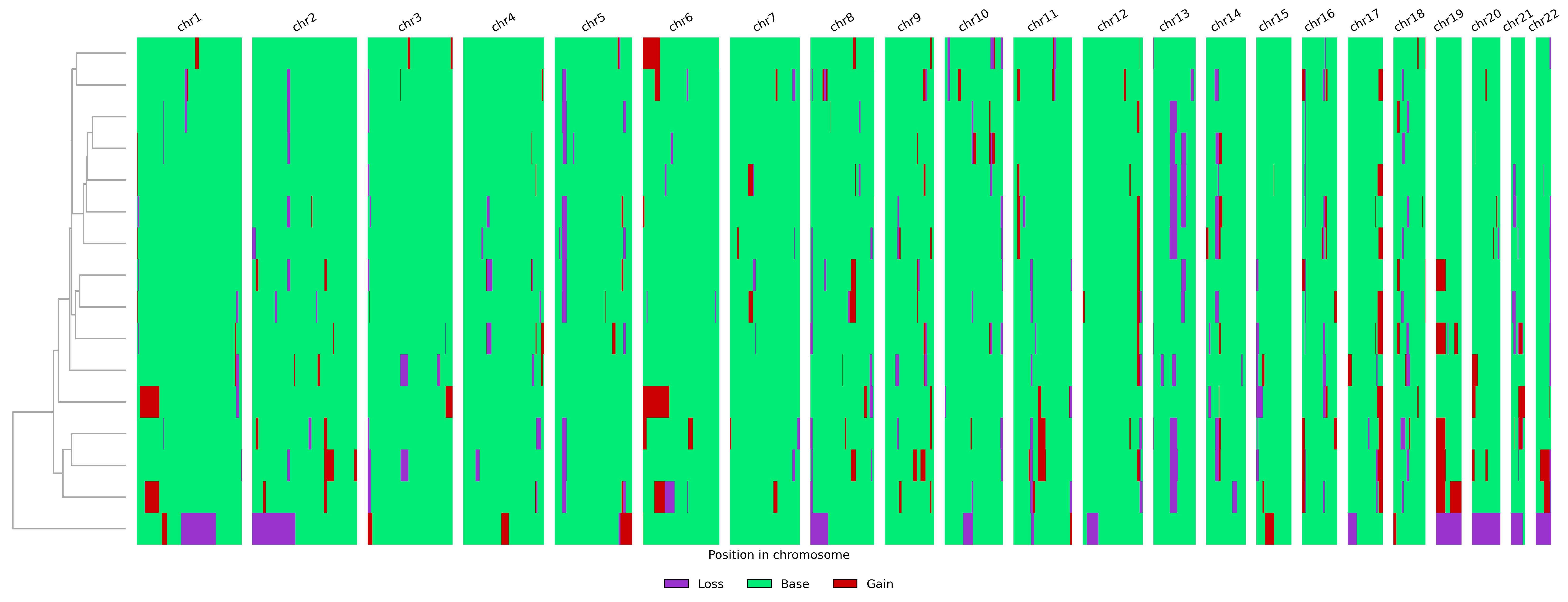
The main output is written to results_sample_data/outs/result_table.tsv.gz.
This table contains the consensus CNV state per cell and genomic bin, encoded
as 0=loss, 0.5=weak loss, 1=base, 1.5=weak gain, and 2=gain.
Additional intermediate outputs are described on the Input and Output
page.
The example run produces the following karyogram: